在医学及其相邻领域,人工智能(AI)的应用正迅速扩展,其辅助系统已广泛应用于基础科学、转化医学和医疗服务等多个领域。这些领域包括医疗管理自动化、辅助诊断和治疗策略。尤其在诊断和治疗方面,众多AI辅助工具已被纳入医疗设备或体外诊断设备的法定范畴。这些符合规定的AI应用受到地方当局的严格监管,这对设备制造商产生了显著的实际和战略影响,并催生了一系列复杂的创新激励机制。哈佛大学商学院助理教授在2022年12月发表了《The Regulation of Medical AI: Policy Approaches, Data, and Innovation Incentives》一文,该文深入探讨了当前医疗器械的监管环境,特别是与AI软件产品相关的法规,并量化分析了AI技术在FDA监管产品中的应用。研究实证部分专注于分析美国市场中AI支持或驱动的医疗设备(AI设备)的特性,包括这些设备的来源、公司类型、国家分布以及安全性概况(基于相关的不良事件和产品召回数据)。最后,该文讨论了监管框架对医疗AI创新激励的潜在影响。本文对该文章的主要内容予以译介。
1 引言
目前,人工智能(AI)在医疗保健领域广泛应用,其涉及范围从基础科学(如蛋白质折叠机理的解析)到转化医学(比如辅助药物发现的算法),以及优化现有数字产品(例如在全基因组测序软件中应用机器学习算法以纠正数据缺失)。最新研究着重强调了AI在医疗服务中的分类应用,特别是深度学习方法如何潜在地革新医疗服务的景象。AI的主要应用领域可分为三大类:(1)医疗管理与行政工作;(2)诊断支持;(3)治疗策略辅助。
表1 人工智能在医疗保健服务中的应用及示例
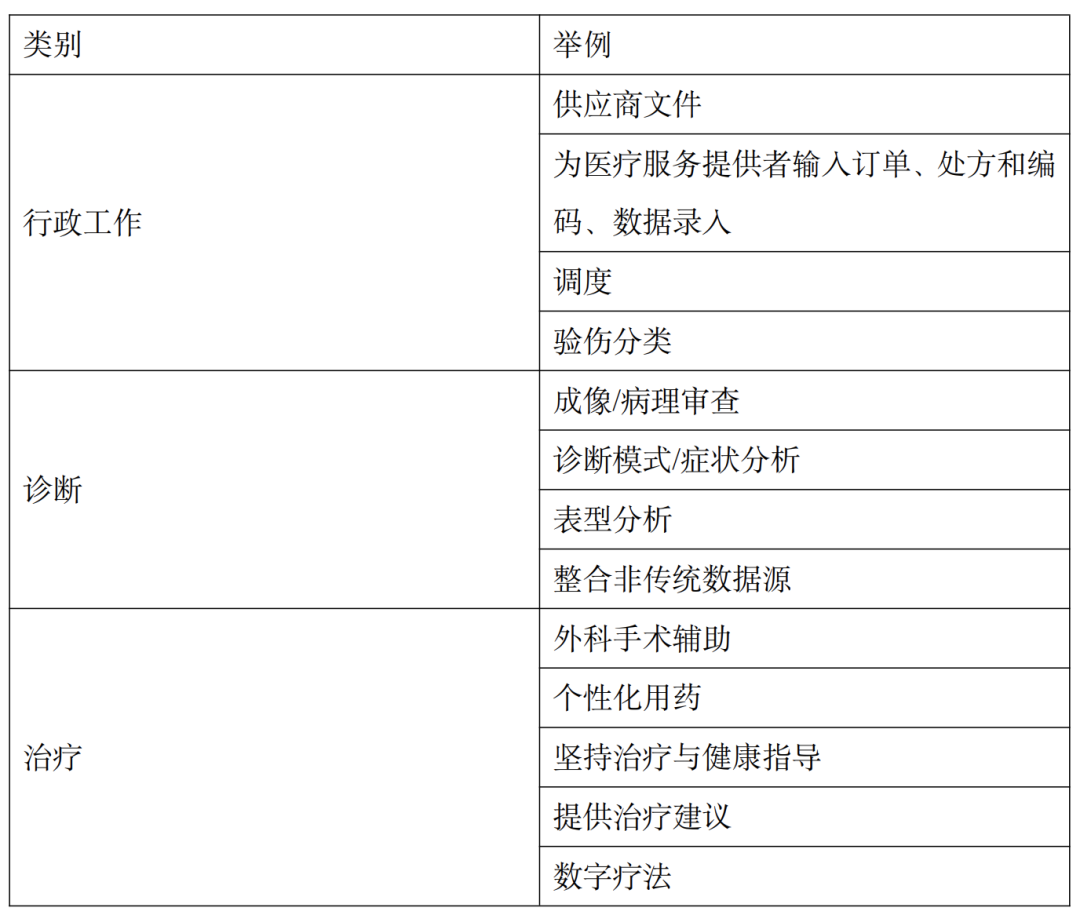
针对常规医疗服务领域,AI在医疗管理工作中的应用需适应医疗服务提供者的特定需求、工作流程和规范(参考Sanders等人,2019年的研究)。除了面临实际操作和设计上的挑战外,这些AI工具还需遵循所在地区的数据隐私法律规定,其中主要包括美国的《健康保险携带与责任法案》(HIPAA)和欧盟的《一般数据保护条例》(GDPR)[1]。AI在医疗管理方面的应用有助于提升医疗服务效率,例如辅助临床医生进行记录存档、日程安排、患者分流、药物订购,及减少用药错误和潜在的药物相互作用。然而,用于行政工作的AI工具往往不符合监管医疗产品的标准,因此在遵守隐私法的同时,它们很少受到医疗设备法规的约束。在诊断和治疗领域,越来越多的AI工具被纳入医疗设备或体外诊断的法定范畴,受到地方监管机构的管控。这对制造商产生了直接影响,同时也引入了更为复杂的创新激励机制。本章将首先介绍美国和欧洲的医疗器械监管背景,尤其关注软件产品的监管挑战,接着深入探讨美国食品与药物管理局(FDA)数据库中登记的受监管AI设备。
本章的实证部分聚焦于美国受监管的、基于软件的、AI支持或驱动的医疗设备。本研究利用公开的医疗器械许可和产品摘要信息,通过文本分析方法识别AI设备,并将其与同类医疗产品进行比较分析。本文详细描述了这些AI设备的特征,包括它们的来源(按公司类型和国别划分),以及基于强制性不良事件报告和产品召回情况的安全性概况。在美国数据的描述性统计分析基础上,本章进一步讨论了监管如何可能塑造AI设备的创新激励,以及监管创新和透明度在受监管AI设备未来发展中可能扮演的角色。最后,本文简要讨论了研究的前瞻性议程。
2 医疗人工智能监管政策办法概览
(一)美国医疗人工智能相关法规
美国的医疗产品法规可追溯到一个多世纪前的1906年《纯净食品和药品法》。然而,现代医疗器械监管始于1976年对1938年《联邦食品、药品和化妆品法》的《医疗器械修正案》(Medical Device Amendments, MDA)。此前,医疗器械一直由各州监管,MDA首次确立了联邦对医疗器械的监管,并建立了当今医疗器械监管的框架。现行医疗器械法规的重点是为用户(包括患者、临床医生、医疗机构和护理人员)提供有关医疗器械安全性和有效性的合理保证。
《联邦食品、药品和化妆品法》第201(h)条将医疗器械定义为由《美国国家处方集》(National Formulary)或《美国药典》(United States Pharmacopoeia)认可的,用于诊断人类或其他动物的疾病或其他状况,或用于治愈、减轻、治疗或预防疾病的仪器、器械、器具、机器、装置、植入物、体外试剂或其他类似或相关物品,包括其组成部分或附件。[2]简而言之,医疗器械是诊断或治疗疾病的工具,而不是代谢(生物或制药)产品。目前,在美国,医疗器械由FDA的器械和放射卫生中心(Center for Devices and Radiological Health, CDRH)监管,美国FDA医疗器械产品目录中共有1700多种。根据风险等级的不同,FDA将医疗器械分为三类(I类、II类、III类),其中45%是I类,47%是第II类,8%是第III类,风险等级逐级升高,III类风险等级最高。具体分类如下:
· I类医疗器械被认为是低风险器械,通常是不含移动部件的简单设计。
适用于I类设备的要求称为“一般控制”。这类器械的示例包括粘性绷带、手术刀和手动听诊器。大多数I类医疗器械可以直接在FDA注册,无需获得许可。
· II类医疗器械属于中风险器械,通常具有更复杂的设计,但如果发生故障还不太可能造成严重伤害或死亡的直接风险。
“一般控制”被认为不足以监管II类医疗器械,因而需要“特殊控制”。II类医疗器械的示例包括内窥镜、电动轮椅、注射器和全关节植入物。有些II类医疗器械只需要注册即可,但更多II类器械需要获得许可。
· III类医疗器械具有高风险,如果发生故障可能导致严重的医疗并发症,甚至死亡。
III类医疗器械需要经过上市前批准(PMA)程序,除非能够证明其与现有获批(“等同”)器械“实质等同”。III类医疗器械的示例包括心脏起搏器、脑深部刺激器、乳房植入物和心脏瓣膜。
所有的器械都必须满足相应的器械的定义和分类监管要求。在美国,全新的器械将会自动归入III类医疗器械,但实际上,很多新产品并没有相当的高风险性。因此,FDA推出“De Novo”注册申请流程。如果FDA通过513(g)或预申请等方式判定器械属于“新型”器械,而没有现有的分类或者市面上不存在等同器械,则可以依据第510(k)条提交De Novo注册申请。在递交De Novo注册申请后的120天内,FDA会判定器械是否属于I类或II类,并签发一个全新的产品代码和监管编号。同时为方便查询器械适用的监管法规,FDA将1700类器械按照医学专业用途分为16类,每类器械均有对应的监管法规条款。除了为新医疗器械建立这些监管途径外,MDA还为在人类患者中进行研究的新研究性器械建立了监管途径——研究性器械豁免,并建立了若干上市后要求和流程,包括不良事件报告要求和良好生产规范。
1990年《安全医疗器械法案》填补了医疗器械监管方面的其他政策空白,授权FDA下令召回器械,对违规行为进行民事处罚,并要求制造商和用户设施(医院、诊所、疗养院等)报告与使用特定医疗器械有关的不良事件,从而改进了上市后的监督。[3]对于软件驱动医疗器械的监管,由于美国医疗器械法规的基础是1976年的MDA,因此在最初的几十年里,有关软件驱动型产品的监管规定并未编入法典。近年来,医疗器械法规才与软件产品的特殊需求和细微差别相匹配。具体来说,医疗器械的任何重大更新历来都需要向监管机构提出新的申请。对于中度风险的器械,没有任何监管规定可以修改或变更现有的510(k)许可,即如果确定修改不在当前510(k)许可范围内,则必须提交新的510(k)。例如,“对临床功能或性能规格有重大影响”的软件变更需要提交新的上市前申请。
目前,在医疗设备中包含软件主要有两种方式。其一,医疗设备可能是软件驱动的,因为它是由与设备功能密不可分的软件驱动的物理设备,有时被称为“医疗设备中的软件”(Software in a Medical Device, SiMD)。SiMD的一个例子是为CT扫描仪提供动力的软件,当没有软件时,硬件设备就无法工作。其二,医疗设备可能完全基于软件,即软件本身符合医疗设备(包括体外诊断)的定义。国际医疗器械监管者论坛将第二类医疗器械称为"作为医疗器械的软件”(Software as a Medical Device, SaMD),并将SaMD定义为用于一种或多种医疗目的的软件,这些软件不作为硬件医疗器械的一部分而实现这些目的",即独立软件(IMDRF,2013年)。
在美国,SaMD产品通常被归类为第二类设备,通过510(k)或De Novo途径进行监管。然而,符合SaMD定义的低风险产品往往有资格获得“执法裁量权”,这意味着FDA不会对这些软件产品执行监管要求。[4]鉴于SaMD产品监管的复杂性,最近有人呼吁监管方法创新,以进一步满足SaMD的独特需求(Torous等,2022年)。
(二)欧盟医疗人工智能相关法规
欧盟的医疗器械监管与美国在基于风险的分类和监管框架方面具有相似性,但也存在重要差异。2017年通过并于2021年5月全面实施的《医疗器械法规2017/745》(MDR)[5]取代了此前的指令和法规。MDR将医疗器械定义为用于一系列医疗目的的各类仪器、器械、软件等。欧盟内的医疗器械需通过合格评定,并获得CE-标志[6]。此外,所有医疗器械必须遵守《一般数据保护条例》(GDPR)[7],以保护个人数据的安全和隐私。Brönneke等人提供了数字医疗产品在欧盟监管、法律和市场环境中的综合概述,包括对MDR和GDPR在数字医疗设备中应用的详细讨论。
3 美国医疗器械的数据监管分析
本节的实证部分探讨了美国受监管的、基于软件的、人工智能支持和人工智能驱动的医疗器械。[8]通过利用有关医疗器械许可和相关产品摘要的详细公开信息,本节利用文本分析来识别人工智能器械,并将其与相同医疗产品领域的其他器械进行比较。
最早对FDA监管的人工智能设备进行调查的研究之一是Benjamens等人的研究,该研究声称发布了“首个全面、开放的数据库,其中包含了FDA批准的严格基于人工智能/移动医疗技术的设备”。该数据库由医学未来学研究所(TMF)负责,截至目前,共包含79种设备。[9]TMF数据库为如何最好地识别人工智能设备,以及哪些医疗专科目前可能与人工智能应用最相关提供了信息。例如,数据库中仅有两款设备通过上市前批准程序对最高风险设备进行监管,其余设备则通过510(k)条或De Novo途径推向市场,这与预认证计划所设想的大多数SaMD产品通过这些途径进入市场是一致的。此外,在TMF数据库中的设备中,80% 以上是放射科或心脏病科设备,这表明这两个医学专业在人工智能设备中的代表性非常高。这些事实有力地表明,将重点放在通过510(k)途径进行监管的设备和最常见的监管医疗专科(包括心脏病学和放射学)上,可以限制数据收集的范围,同时仍有可能管控绝大多数受FDA监管的人工智能设备。美国食品和药物管理局随后公布了一份人工智能设备清单,并于2022年10月进行了更新,证实了类似的模式,自2010年以来上市的数百种人工智能设备中,只有两种是通过PMA途径上市的。[10]基于此,本节接下来的数据收集策略和实证分析将基于以数据为依据的方法。
作为分析的基础,本节下载并分析了2010年至2022年第三季度的完整的510(k)数据[11],并重点关注了八个最大的医疗分类(由各自的FDA咨询委员会小组定义),如表2所示:(1) 临床化学和毒理学器械;(2) 心血管器械;(3) 牙科产品;(4) 消化内科-泌尿科器械;(5) 综合医院和个人使用器械;(6) 整形外科和康复器械;(7) 放射科器械;以及 (8) 普通外科和整形外科器械。
510(k)数据库包括有关器械类型(产品代码)的信息、有关申请公司(器械制造商)的数据(例如其名称和申请地址)、向监管机构提交每份申请的日期、每种器械获得 FDA 批准的日期,以及与器械相关的医学专业(分配给FDA医疗器械咨询委员会之一[12])。此外,本文将有关器械结果的另外两个信息来源合并到该数据库中:(1) FDA的医疗器械不良事件报告数据库(MAUDE数据库[13])中收集的与医疗器械相关的不良事件数据;(2) FDA的召回数据库中有关医疗器械召回的数据[14]。
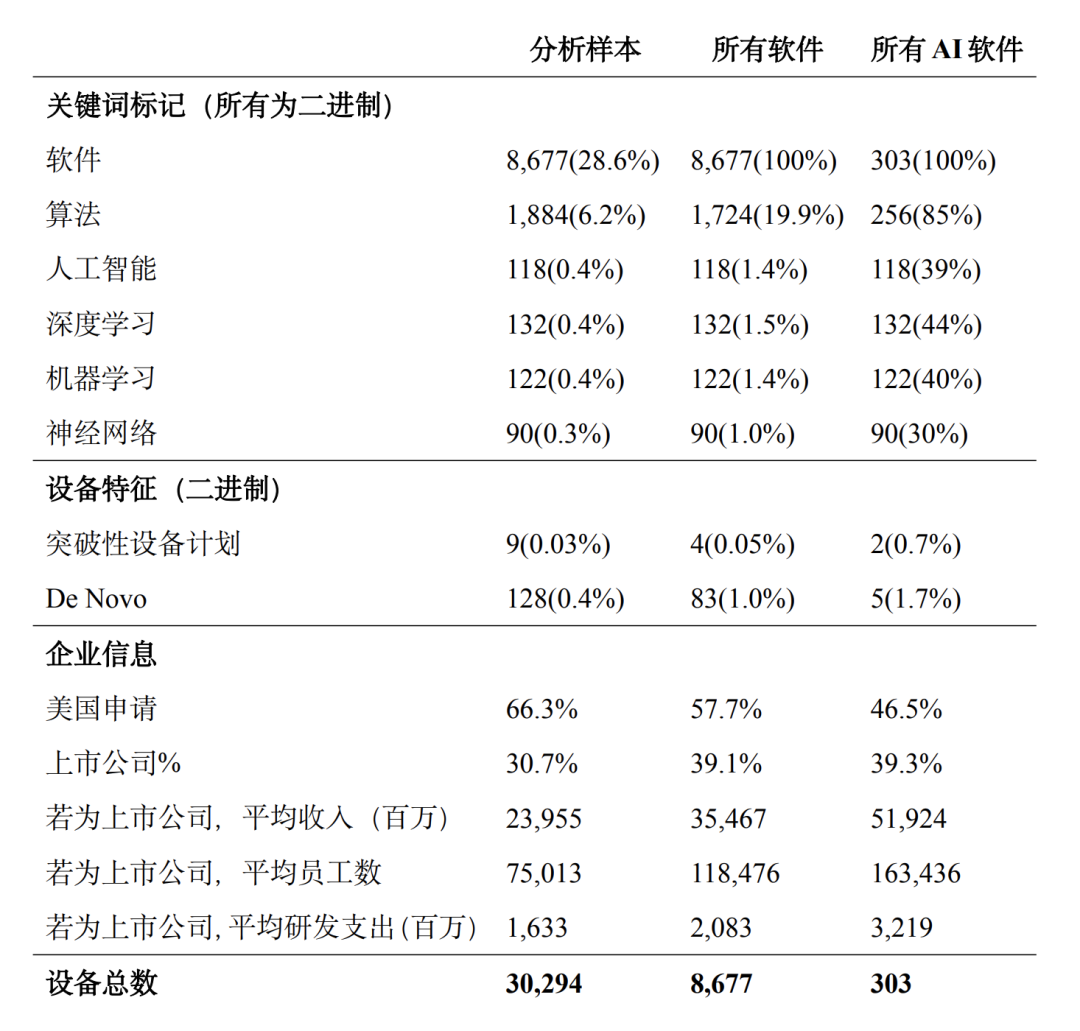
接下来的步骤将使用文本分析来识别具有软件组件的设备以及包含人工智能的设备(即上文定义的“人工智能设备”)。这两项工作都依赖于机器可读的公开摘要文件。在我们分析的30,779种前8位专业设备中,30,294种(98.4%)有此类文档,这些文档构成了用于标记相关设备的文本分析的基础。本文采用Stern和Foroughi描述的算法来识别所有软件设备(包括SiMD和SaMD)[15],然后进行进一步的关键词搜索,将人工智能设备识别为其中的一个子集。为这项工作特别选择的关键词是“人工智能”、“深度学习”、“机器学习”和“神经网络”,选择这些术语是因为它们与人工智能算法的描述有直接关系,而且在使用时可能没有歧义。[16]其中,图1是分析样本构建流程图,表2是按医学专业划分的分析样本明细。
图1: 分析样本构建流程图
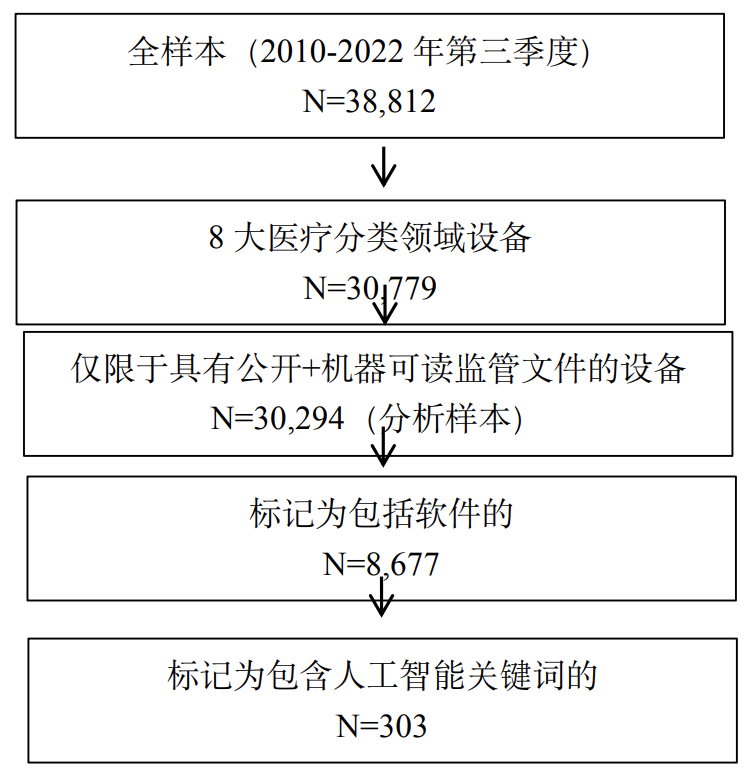
表2: 按分类分列的设备分析样本
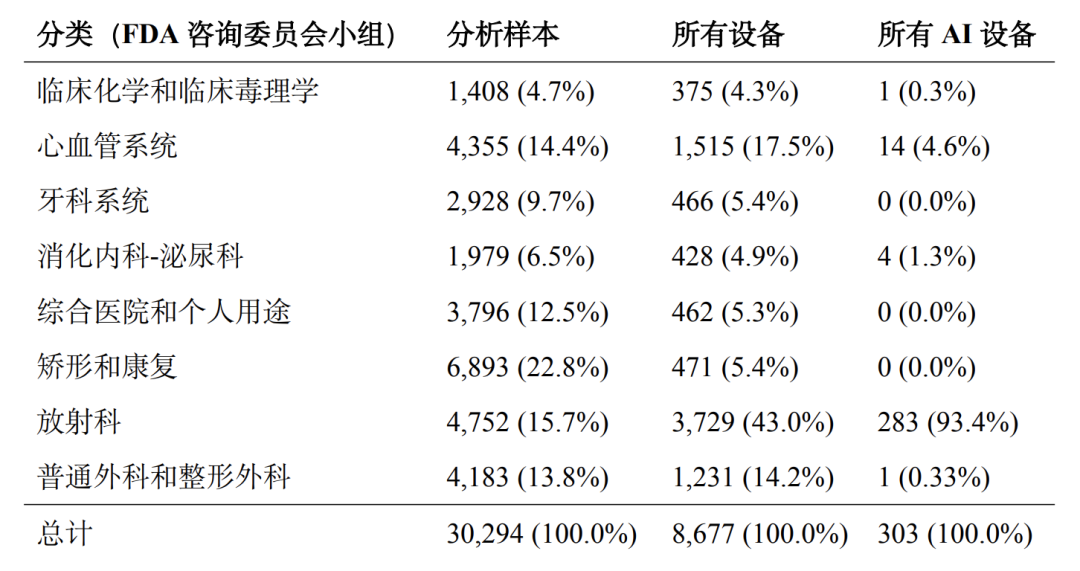
从分析样本的汇总统计数据(表3)中可以看出,首先,近29%的分析样本设备包含软件。这与Stern和 Foroughi的观点一致,他们记录了医疗设备行业的显著数字化,其中放射科设备的SiMD和SaMD率最高,其次是心脏科设备。当然,包含软件组件的数字化设备是融入人工智能的必要条件,但并非充分条件。
表3: 分析样本的汇总统计数据
4 医疗人工智能的创新激励措施
在医疗AI与医疗产品法规交汇处,创新政策研究者应如何思考?从美国对受规管AI的初步数据中我们能学到什么?回答这些问题的一个明确起点是了解FDA当前对AI/ML产品的监管立场,这一立场体现在2021年初发布的《AI/ML软件作为医疗设备的行动计划》(以下简称为《行动计划》),以及之前发布的“AI/ML基础软件作为医疗设备(SaMD)修改的拟议监管框架——讨论文件及反馈请求”中。这些文件中的观点基于国际医疗器械监管论坛(IMDRF)所概述的风险分类原则,并以软件预认证试点计划所构想的“全生命周期方法”为基础。
对AI/ML设备未来监管的拟议框架中,一个关键部分是“算法变更协议”。这个协议将依赖于现实世界的性能数据和持续监测,使监管机构能够灵活地平衡FDA的双重任务:保护公众健康,同时又能及时提供新产品。更广泛地说,《行动计划》概述了FDA未来工作的五个关键方向:
· 为基于AI/ML的SaMD制定特定的监管框架
· 促进良好机器学习实践(GMLP)的发展和标准化
· 采取以患者为中心、增加用户透明度的方法
· 使用与算法偏差和鲁棒性有关的监管科学方法
· 现实世界的性能表现
《行动计划》的首要目标是为基于AI/ML的SaMD建立特定的监管框架,这清楚地表明美国监管机构已意识到监管基于AI的SaMD所固有的特殊挑战和机遇。然而,如何精确实施AI设备法规在实践中仍然存在很大的不确定性。此外,《行动计划》的各个部分提出了许多未解决的问题:例如,即将出现的特定监管框架的范围和具体性如何?CDRH计划鼓励GMLP的协调,预计包括数据管理、特征提取、训练、可解释性评估和文件记录等最佳实践,FDA承认这些与良好软件工程实践或质量体系实践相似——这些GMLP将具有多少细节和负担?它们将如何与算法开发的最新状态保持一致?这些问题的答案,尤其是后者,将显著影响开发者进入受监管的SaMD领域的障碍。此外,FDA还与国际标准化组织和英国标准协会等在AI医疗技术方面的国际组织保持合作关系。这些合作关系非常重要:国际公认的标准设定机构对于识别最有前途的技术和影响技术采纳轨迹至关重要,而且,监管明确性被认为可以加快其他类型医疗设备的上市时间。
在以患者为中心的用户透明度、与算法偏差和鲁棒性有关的监管科学方法,以及对现实世界性能数据的创造性但严格的应用方面,仍有许多类似性质的问题需要由利益相关者提出并由监管机构回答。AI开发的一个挑战通常是缺乏透明度,即AI/ML算法如神经网络是如何执行其任务的;目前这是不可能的。尽管如此,仍需要通过特定的验证测试来建立对结果的信任,这正是“现实世界性能”研究的重要作用所在。在所有这些主题上,更广泛的数字医学社区也可以共同努力,推动医疗AI的发展,例如在生成和使用数字健康产品评估的实际证据方面制定最佳实践和优先事项。这项工作将涉及以患者为中心的透明度和监管科学的多个方面。
最后,《行动计划》范围之外的其他问题也迅速浮现:如何处理定义在此的大量AI设备(SiMD而不是SaMD产品)?行动计划的范围明确限于独立软件,这意味着对AI SiMD产品可能仍存在很多不确定性。在这种背景下,与监管不确定性概念相关的创新政策问题变得更加尖锐。在美国医疗设备环境中,监管不确定性——例如,当首创的医疗设备首次经历监管审批过程时——与首次进入市场的劣势和小公司新颖设备商业化率较低有关。有理由相信,监管不确定性将负面影响公司参与受监管空间的创新意愿,特别是那些没有监管先例的更具创新性的研发项目。但这也表明,积极的方法在这种情况下可能具有巨大的潜力:通过正式指导文件提供监管明确性已被证明可以加快新的高风险设备的监管批准,这表明监管创新和政策澄清在塑造创新激励方面的重要作用。
综上所述,在AI设备背景下,监管创新和监管明确性的价值可能特别重要,因为迄今为止的大部分创新都来自于小型公司和其他国家的公司。大型(例如上市)和国内公司更有可能首先拥有美国监管专业知识,并且这些公司平均在(美国)受监管的产品空间中比它们的小型和/或国际基础同行更有经验。然而,美国AI设备推出的早期证据表明,私人持有的国际制造商开发AI设备的可能性与在受监管空间中看到的其他情况相比不成比例。
5 结论
本研究对受监管的医疗器械领域进行的调查揭示了一个关键趋势:近年来,人工智能(AI)在医疗产品方面的商业化程度急剧上升。监管机构,尤其是美国食品及药物管理局(FDA),已开始深入探讨如何对这类技术进行有效监管,旨在同时确保公众健康和促进患者获取创新医疗技术的便利。在此过程中,创新激励措施在多个方面发挥着重要作用。人工智能在医疗诊断和治疗应用方面的广泛性意味着许多此类工具可能会被归类为正式的医疗设备。因此,医疗产品监管不仅继续在此领域发挥作用,而且对软件开发商、成熟的医疗设备公司、风险资本投资者以及创新用户形成了显著的激励。
人工智能工具是否符合医疗设备的标准,以及这一界定在多大程度上存在模糊地带,都对产品的开发、研发决策以及其在医疗系统中的商业化和支付方式产生重要影响。在受监管的医疗器械领域,根据器械被归类为低、中或高风险,所需的临床证据和文书工作负担之间存在显著差异。特别是对于AI开发人员而言,这些差异可能更加突出,因为他们相比于传统医疗技术公司的研发人员,可能缺乏足够的医疗器械监管经验。美国食品和药物管理局最近发布的指导方针,以及数字医学界更广泛的最佳实践(包括临床研究人员、标准制定组织和公司),都将对新产品开发的不确定性产生影响,从而激励各大小公司在医疗人工智能领域进行创新。
[1] The European Union’s General Data Protection Regulation (GDPR): https://gdpr-info.eu/art-1-gdpr
[2] https://www.fda.gov/med%EF%BF%BEical-devices/premarket-submissions-selecting-and-preparing-correct-submission/premarket-notification-510k
[3] https://www.fda.gov/medical-devices/how-study-and-market-your-device/breakthrough-devices-program
[4] https://www.fda.gov/media/80958/download; Other sometimes-regulated products include “smart wearables” such as devices manufactured by Fitbit, AliveCor, Grarmin, and Apple, which have both medical and consumer applications. Felber and Maciorowski (2023) provide an overview of how such “smart wearables” are used and regulated in the United States along with relevant risks and public purpose considerations.
[5] https://eur-lex.europa.eu/eli/reg/2017/745/2020-04-24
[6] https://single-market-economy.ec.europa.eu/single-market/goods/building-blocks/notified-bodies_en
[7] https://gdpr-info.eu/
[8] This study does not distinguish between AI-supported vs. (entirely) AI-driven medical devices. For example, a piece of radiology equipment that uses AI to improve image quality (AI-supported) would qualify as an AI device, as would a SaMD product in which the algorithm itself constitutes the entirety of the medical device (fully AI-driven).
[9] 2 List pulled on June 7, 2022 from https://medicalfuturist.com/fda-approved-ai-based-algorithms
[10] For the most recent list of AI/ML devices from the FDA see https://www.fda.gov/medical-devices/software-medical-device-samd/artificialintelligence-and-machine-learning-aiml-enabled-medical-devices
[11] Data downloaded on October 4, 2002 from https://www.fda.gov/medical-devices/510k-clearances/downloadable-510k-files
[12] https://www.fda.gov/advisory-committees/medical-devices/medical-devices-advisory-committee
[13] Data downloaded on March 2, 2022 from https://www.fda.gov/medical-devices/mandatory-reporting-requirements-manufacturers-import
ers-and-device-user-facilities/manufacturer-and-user-facility-device-experience-database-maude
[14] Manually downloaded and scraped recall database from FDA in May 2021 https://www.ac
cessdata.fda.gov/scripts/cdrh/cfdocs/cfres/res.cfm
[15] Stern, Ariel Dora. “Innovation under regulatory uncertainty: Evidence from medical technology.” Journal of public economics 145 (2017): 181-200.; Foroughi, Cirrus, and Ariel Dora Stern. “Who Drives Digital Innovation?: Evidence from the US Medical Device Industry.” Harvard Business School Working Paper, 2019.
[16] 人工检查设备摘要的随机样本证实,根据这种方法,误报率为 0%
撰稿 | 仇怡然,清华大学智能法治研究院实习生
修改、指导 | 鲍伊帆、刘云
编辑 | 朱正熙
注:本公众号原创文章的著作权均归属于清华大学智能法治研究院,需转载者请在本公众号后台留言或者发送申请至computational_law@tsinghua.edu.cn,申请需注明拟转载公众号/网站名称、主理者基本信息、拟转载的文章标题等基本信息。
声明:本文来自清华大学智能法治研究院,版权归作者所有。文章内容仅代表作者独立观点,不代表安全内参立场,转载目的在于传递更多信息。如有侵权,请联系 anquanneican@163.com。
